
Dr. Siddharth Madan, S16243, Dr. Zia Chaudhuri, Dr. Zia Chaudhuri, Dr. Om Prakash
Abstract
A 4-year boy presented with mild right upper eyelid ptosis following direct ocular trauma with a cricket bat. Ocular movements were full with good levator action. Post-traumatic ptosis was suspected and patient was advised observation. 6 months later, he had moderately severe ptosis both eyes, ophthalmoplegia, bilateral frontalis overaction, chin up position and no significant refractive error. Both eyes had symmetrical outward deviation from midline and ocular movements were limited in all gazes with minimally preserved lateral movements in the right eye. A retrospective history of diurnal variation of ptosis prompted anti-acetylcholine receptor antibody level assay, which were significantly raised. The neostigmine stimulation test was positive. CT chest was normal and no thymoma was seen. The child improved markedly with 15 mg pyridostigmine. OMG should be suspected in any acquired symmetrical ophthalmoplegia not conforming to a neural/restrictive pattern of strabismus.
Key words: Juvenile myasthenic ophthalmoplegia, Ocular myasthenia gravis
Synopsis: Ocular myasthenia gravis (OMG) should be suspected in any acquired ophthalmoplegia not conforming to a neural / restrictive pattern of strabismus.OMG is not a life-threatening condition but requires life long medical support. A vast majority of patients with OMG progress to generalized myasthenia, which is a life-threatening condition.
Introduction
Myasthenia gravis (MG) is a post synaptic autoimmune disorder that leads to dysfunction of acetylcholine receptors. Ocular myasthenia gravis (OMG) is a subtype of MG. Onset of the disease in the first decade or after the age of 70 years is less commonly observed.1More males are affected by pure OMG. The incidence of OMG is higher among children, particularly oriental children.2 Weakness is clinically localised to extraocular muscles (EOM), levator palpebrae superioris (LPS) and orbicularis oculi muscles that manifests with eyelid ptosis and diplopia in nearly three fourth of the patients.3
Case Report
A 4-year old male child was hit by cricket bat on face while playing 6 months prior to presentation to us. He developed swelling over right side of forehead above eye. However, there was no accompanying history of any direct injury to the eye, redness and watering or associated diminution of vision. After 15 days, mother noticed mild drooping of right upper lid. As per his clinical records of a consultation elsewhere, the documented LPS action was good, ocular movements were full in all directions of gaze and he was advised observation for his complaints on suspecting a diagnosis of post traumatic ptosis. 6 months later he presented to the ophthalmic unit of our pediatric hospital with progressively increasing drooping of right upper eyelid (Fig. 1A). Left upper eyelid droop was noted a month before presentation (Fig. 1A).The parents did not appreciate any history of deviation of eyes. There was no history of recent immunization, spectacle use, ocular surgery, swallowing difficulty or any other significant past (Fig.1B) or family history. On examination, the child could recognise snellen’s picture chart corresponding to a visual acuity of 6/12 in either eye. Pupillary reactions were normal and both the visual axis deviated outwards from the midline (Fig. 1G). There was an apparent exotropia of nearly 40 prism diopters in the right eye in primary gaze. Ocular movements were limited in all gazes except for a preserved abduction and nearly normal dextroelevation and dextrodepression in the right eye (Fig. 1C, 1F, 1I). Upper eyelid crease was absent bilaterally. Ptosis was severe in the right eye and moderate to severe in the left eye. LPS action (4 mm in right eye and 5 mm in left eye) and Bell’s phenomenon was poor in both the eyes. He had chin up position with overacting bilateral frontalis muscles (Fig. 1A). Refraction under atropine revealed a simple myopic astigmatism with the rule of -0.50 and -1.00 diopter cylinder in right and left eye respectively which was prescribed to the patient.
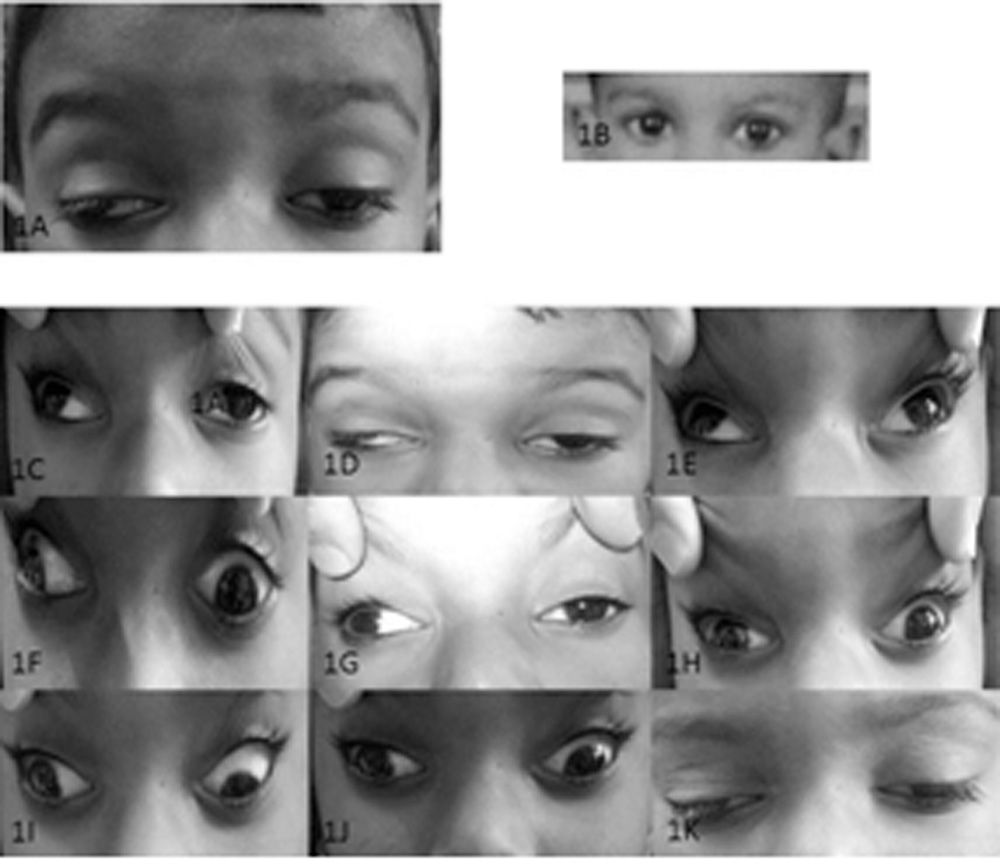
A 4-year old male child presented with severe ptosis OU (Fig. 1A, 1G). He was apparently well in the past with no ptosis or ophthalmoplegia (Fig. 1B). There was exotropia of 40 prism diopters in primary gaze (Fig. 1G). Ocular movements were limited in all gazes (Fig.1D, 1E, 1H, 1J, 1K) except for a preserved abduction (Fig. 1F) and nearly normal dextroelevation (Fig. 1C) and dextrodepression (Fig. 1I).
Rest of the anterior, posterior segment and fundus examination was normal (Fig.2A, 2B). Suspecting a possibility of juvenile OMG and chronic progressive external ophthalmoplegia, antibodies to the acetylcholine receptor (AChR Abs) were advised which were found to be0.43 nmol/l and were markedly raised (normal range is < 0.25 nmol/l ). Computed tomography and antero-posterior X-ray of the chest was normal without any evidence of a thymoma (Fig. 2C, 2D).
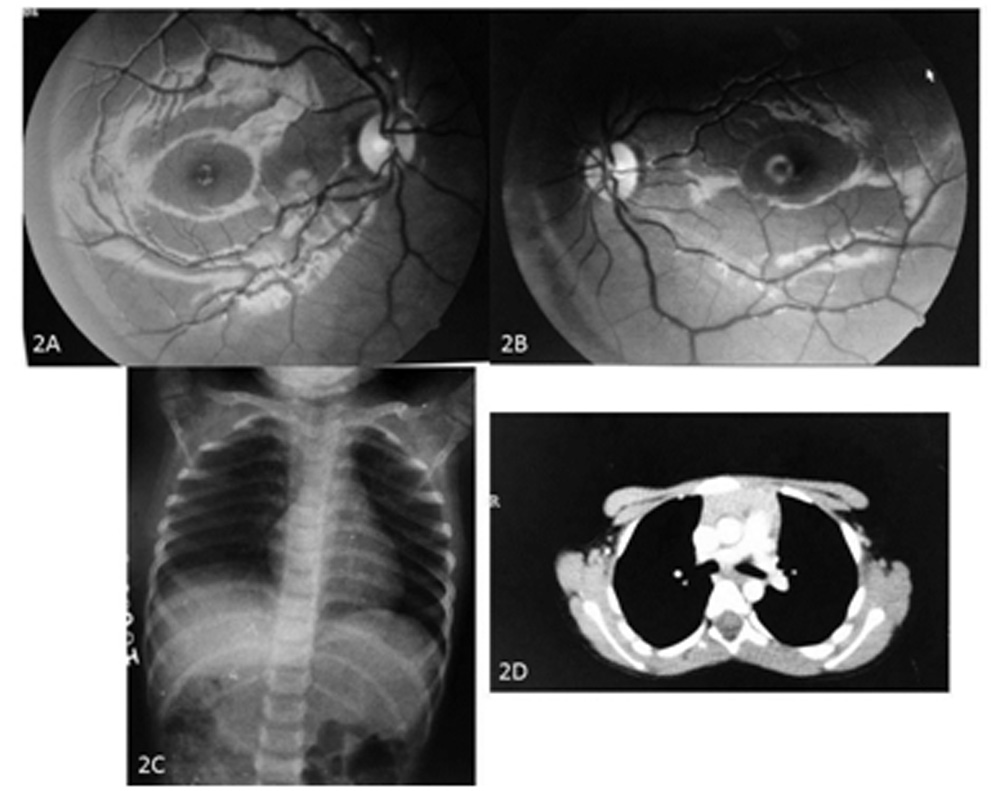
Figure 2A-2D: Fundus photographs and Imaging: Fundus examination was normal OU (Fig. 2A, B). Chest X ray (antero-posterior) view (Fig. 2C) and CT chest (Fig. 2D) was normal. No thymoma was seen.
Neostigmine stimulation test after injecting 1.5 mg intramuscular neostigmine (acetylcholinesterase inhibitor) was positive with a marked improvement in ptosis (Fig. 3A, 3B). He was additionally diagnosed with sub-clinical hypothyroidism with elevated thyroid stimulating hormone (TSH) levels (7.07 mIU/L) but normal T3 and T4 levels. Anti-TSH antibodies were negative. The child was started on 15 mg pyridostigmine 6 hourly and demonstrated significant improvement in ptosis without much improvement in ophthalmoplegia after a follow-up of 1 month (Fig. 3C).
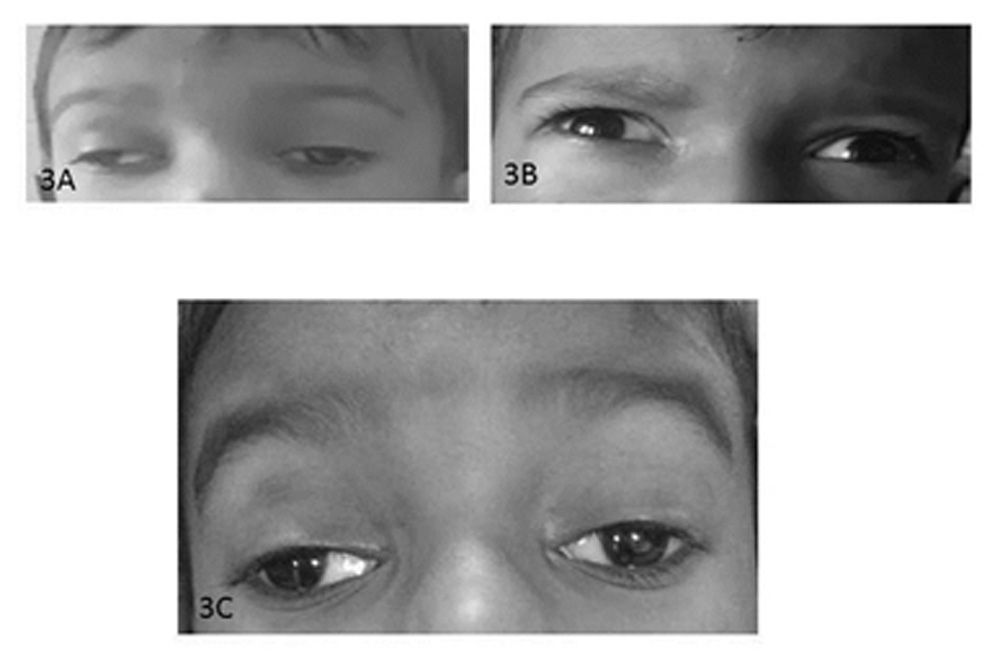
Figure 3A-3C: Neostigmine stimulation test and post-treatment photograph: Neostigmine stimulation test after injecting 1.5 mg intramuscular neostigmine (acetylcholinesterase inhibitor) was positive with a marked improvement in ptosis (Fig.3B) as compared to pre-administration of the drug (Fig 3A). A significant improvement in ptosis without much improvement in ophthalmoplegia was noticed after initiation of treatment at a follow-up of 1 month (Fig. 3C).
Discussion
Childhood MG can be classified into transient neonatal myasthenia gravis and juvenile myastheniagravis (JMG).4 Transient neonatal MG occurs in about 10% of babies as result of transplacental transfer of maternal (AChR) and manifest with hypotonia and feeding difficulties. Resolution may occur within two months primarily with supportive treatment.4JMGoccurs as a result of AChR Abs and muscle-specific kinase (MuSK) that leads to decreased AChRactivity through complement mediated lysis of the post-synaptic membrane and therefore presents with fatigability.5It is seen before the age of 19 and in contrast to the congenital myasthenic syndromes (CMS), is not due to a structural abnormality in the neuromuscular junction.6CMS affected patients may have a turbulent post-natal period and typically present before two years of age closely mimicking MG, posing diagnostic difficulties.
In around 20% of JMG patients, symptoms remain limited to EOM and almost all patients experience some degree of ocular weakness during the course of their disease.3Ptosis is mostly asymmetrical and may be alternating. Severe ptosis can be a risk for developing amblyopia. Diplopia may occur due to weakness of a single muscle however, bilateral pareses are common. Hallmark of this disease remains fatigability and fluctuating ophthalmic signs. Nearly 90% of all children with JMG will have ocular symptoms and about 50% consult an ophthalmologist first.4
OMG can pose a diagnostic dilemma and can clinically resemble isolated cranial nerve palsies, gaze palsies, internuclear ophthalmoplegia, blepharospasm, stroke to even complete ophthalmoplegia.7A strong clinical suspicion is required in any variable incomitant strabismus, with or without the presence of ptosis.Acute incomitant strabismus or external ophthalmoplegia is the second most common presentation of the disease after ptosis and most of these children may be misdiagnosed as having intracranial tumors.8Medial rectus is most commonly involved.8Pupils are usually uninvolved and helps distinguish the disease clinically from partial third nerve palsy, Horner’s syndrome and Botulism. Mild or relatively well-controlled myasthenic children may demonstrate detectable muscle fatigue on physical examination upon fatiguingLPS with sustained voluntary upgaze for at least a minute (Cogan twitch sign) as observed in this case. Diagnosis of OMG is chiefly clinical.Between 50 to 80 % of OMG progress to general myastheniagravis (GMG)with involvement of other muscles and presents with limb weakness and bulbar symptoms within two years.2 Hashimoto’s disease and autoimmune polymyositis may be seen concomitantly.9
AChR Abs are detected in 50% of patients with ocular myasthenia and in 80–85% of those with generalised disease by radio-immunoassay (RIA) or cell-based assay.10A positiveAChR Ab test is virtually diagnostic in the appropriate clinical setting due to rare false positive rate.10-15%of patients who are negative for AChR Abs may demonstrate antibodies to MuSK or the low-density lipoprotein receptor protein 4 (Lrp4).10 Patients positive for MuSK antibodies have less observable clinical fluctuations and have an increased predisposition to myopathic weakness.
Edrophonium testing using intravenous administration of edrophonium, a short acting cholinesterase inhibitor helps diagnose OMG.2, 3,4, 5,7 A positive test indicates a rapid but transient improvement of the clinical sign, most commonly ptosis and occasionally dysarthria or ophthalmoplegia. A small yet a definite risk of cardiorespiratory collapse, bradycardia, and excess salivation necessitateexecution of this test under monitoring.
OMG is not a life-threatening but a lifelong disease. Available treatment options are currently to improvevision, are supportive and to prevent progression to GMG. The use of acetylcholinesterase inhibitors chiefly pyridostigmine is disease specific.2, 3, 4, 5, 7 Patients with GMG require a much higher degree of immunosuppression than those with OMG, using prednisone, azathioprine, and plasmapheresis or intravenous immunoglobulin.2, 3, 4, 5, 7 Thymectomy may be considered in children with suspected thymoma yet the treatment mostly remains conservative.4
References
- O’Brien MD. The miracle at St Alfeges: Seventy years on. J R Soc Med 2007;100:257.
- Nair AG, Patil-Chhablani P, Venkatramani DV, Gandhi RA. Ocular myasthenia gravis: A review. Indian J Ophthalmol 2014;62:985-91.
- Mullaney P, Vajsar J, Smith R, Buncic JR. The natural history and ophthalmic involvement in childhood myasthenia gravis at the hospital for sick children. Ophthalmology 2000; 107:504–510
- Liew WKM, Kang PB. Update on juvenile myasthenia gravis. Curr Opin Pediatr 2013;25:694-700.
- Evoli A. Acquired myasthenia gravis in childhood. Curr Opin Neurol 2010;23:536-40.
- Kusner LL, Puwanant A, Kaminski HJ. Ocular myasthenia: diagnosis, treatment, and pathogenesis. Neurologist 2006;12:231–9
- Sommer N, Melms A, Weller M, Dichgans J. Ocular myasthenia gravis. A critical review of clinical and pathophysiological aspects. Doc Ophthalmol 1993;84:309‑33
- Berkovitch S, Belkin M, Tenenbaum A. Childhood myasthenia gravis. J Pediatr Ophthalmol 1977;14:269 –73
- Tsao C, Mendell JR, Lo WD, Luquette M, Rennebohm R. Myasthenia gravis and associated autoimmune diseases in childhood. J Child Neurol 2000;15:767-9.
- Evoli A, et al. J Neurol Neurosurg Psychiatry 2017;0:1–3. doi:10.1136/jnnp-2017-315782
Leave a Comment