
Dr. Anupama Janardhanan, A20042, Dr. Mary Varghese
ABSTRACT
Aim: To describe the clinical picture and visual outcome in patients with NMO.
Method: Retrospective analysis of 32 eyes of 16 patients with IgG positive NMO.
Results: NMO was mainly seen in female patients (75%). Visual loss was seen in 25 of 32 eyes with bilateral involvement in 75% and associated neurological symptoms in 88% of patients. BCVA at time of presentation was between Perception of light (PL) to 2.0 (LogMAR) in 32%, 1.3-1.77 in 40%, 0.77-0.0 in 24%. Papillitis was seen in 17 of 25 eyes had while 8 had optic atrophy. VEP showed prolonged P100 latencies in 82%. All patients received IV Methylprednisolone 1gm for 5 days followed by oral steroids. BCVA after treatment showed 2 lines improvement in 24%, 4 lines improvement in 32% and upto 6 lines in 20%.
Conclusion: NMO was most commonly seen in female patients. NMO IgG test may be advisable in patients with papillitis with neurological symptoms. Prompt diagnosis and early treatment with systemic steroids may lead to good visual recovery.
INTRODUCTION
Neuromyelitis optica / Devic’s Disease is a unique astrocytopathy. It is an antibody-mediated, inflammatory disease of the central nervous system mainly affecting the optic nerves, spinal cord and peri-ependymal regions of the cerebral hemispheres and brainstem.1 It is basically characterized by longitudinally extensive transverse myelitis and severe attacks of optic neuritis (ON).2 It can have a monophasic or a relapsing course. The functional impairment during an episode can be devastating with no significant improvement in vision or motor strength in some patients. Patients diagnosed for the first time with optic neuritis or myelitis often pose a question whether the event will recur or predicts limited NMO or it is a clinically isolated syndrome of future MS.3
Earlier, NMO was thought to be a subtype of multiple sclerosis (MS). Recent advances especially discovery of NMO-IgG, an NMO-specific autoantibody directed against aquaporin-4 (AQP4), the major water channel in the central nervous system has delineated NMO as a separate disease from MS.4 Differentiating these conditions is of prime importance because early initiation of immunosuppressive therapy is the only effective way of preventing attack-related disability in NMO spectrum disorders (NMOSD). Another specific reason being that certain drugs for multiple sclerosis may exacerbate NMOSD.2
Visual impairment in NMO is severe with poor recovery if untreated. 5 NMO attacks are incapacitating with about 50% of those diagnosed remaining dependent on a wheelchair and functionally blind by 5 years of diagnosis. 6
METHOD
This is a retrospective study of 32 eyes of 16 patients with IgG positive NMO who were admitted in our tertiary hospital from April 2015- April 2017. In this study we evaluated the demography, clinical features, visual involvement and outcome following treatment at our hospital.
Clinical evaluation included visual acuity, pupillary reaction, presence of relative afferent pupillary defect, fundus examination and neurological examination. All patients underwent MRI scans of Brain / Spinal cord and Orbits as well as visual evoked potential (VEP).
Blood investigations confirming the positivity of NMO IgG antibody constituted the main inclusion criterion for the study. All patients received IV Methylprednisolone 500 mg twice daily for 5 days followed by oral steroids in tapering doses A few patients also received specific monoclonal antibody in form of Rituximab.
STATISTICAL ANALYSIS
Descriptive statistics including mean and standard deviation for continuous variables and frequency counts with percentages were computed.
RESULTS
Complete details of 16 patients were available and included in the analysis.
Immunofluorscence (IF) based NMO IgG antibody testing was done for all suspected NMO cases. Only NMO IgG positive cases diagnosed as per Wingerchuk’s revised criteria were included in this study 10.
The 16 patients were between the ages of 25 to 84 years with a mean age of 39.1 +/- 9.4 years {Table-1}. There were 12 females (75%) and14 of the 16 patients had associated neurological involvement.
14 of the 16 (87 %) patients had ocular complaints in form of acute visual loss. Four patients showed unilateral ocular involvement whereas 12 patients had bilateral involvement at the time of analysis, either as an acute manifestation or as a result of prior effect. 25 out of 32 eyes were involved and the remaining eyes were normal at the time of examination.
BCVA at time of presentation was between Perception of Light (PL) to 2.0 (LogMAR) in 32%, 1.3-1.77 in 40%, 0.77-0.0 in 24% {Table-2}.
On fundus examination 17 of 25 eyes (68%) had acute papillitis while 8 eyes (32%) had clinically evident optic atrophy. All patients underwent VEP as a part of evaluation at the time of presentation. VEP showed prolonged P100 latencies in 82% of the total number of eyes (32 eyes) evaluated.
CSF analysis was done for all patients to rule out infective causes for neurological symptoms. All patients underwent MRI Brain +/- Spine depending on type of presentation of NMOSD. 12 out of 16 patients evaluated showed MRI findings falling within the domain of Imaging criteria for NMOSD as per Wingerchuk 10.
All patients received IV Methylprednisolone 1gm for 5 days followed by oral steroids. 3 out of 16 patients received Rituximab along with maintenance low dose oral steroids. BCVA after treatment showed 2 lines improvement in 24%, 4 lines improvement in 32% and upto 6 lines in 20% of total patients with ocular involvement. {Table-3}
80 % patients showed improvement in systemic symptoms within 1 week of initiation of intravenous corticosteroids.
DISCUSSION
Neuromyelitis optica spectrum disorder is diagnosed on the basis of Wingerchuk 2015 Diagnostic Criteria for NMOSD10. It classified clinical features into Absolute core presentations; optic neuritis (bilateral/ unilateral) or acute myelitis (> 3 contiguous cord segments), area postrema syndrome, acute brainstem syndrome, diencephalic syndrome or cerebral syndrome and Supportive presentations including; Positive NMO specific IgG autoantibody (Ab), LETM on T2 weighted MRI, Non specific MRI Brain lesions.4
Seropositive NMO defined as having one of the absolute core presentation with positive NMO IgG Ab. Seronegative NMO requires criteria of two absolute core presentations with additional specified imaging criteria6. Only seropositive NMO cases were included in the study.
The incidence of NMOSD ranges from 0.053 to 0.4 per 100,000 individuals. 1
NMO commonly manifests for the first time around the fourth decade of life
with female preponderance5. 75% of affected patients in our study were females.
Aquaporin-4 (AQP4) is one of the main water channels in the CNS. It is a bidirectional channel which is highly expressed in astrocytic foot processes and is critical for water, glutamate and potassium transport.7
In 2004, Lennon et al reported presence of NMO-IgG. The target antigen of NMO-IgG was subsequently confirmed to be AQP4. 5
Expression of AQP4 in the optic nerves and spinal cord and a deficiency of tight junctions between endothelial cells forming a permeable blood-brain barrier in these areas make them more prone for IgG antibody attack..1
In vitro studies show that binding of NMO-IgG to AQP4 initiates multiple pathogenic mechanisms. AQP4 down-regulation occurs through antigenic modulation leading to targeting of antigen to lysosomal pathways. Finally activating the classical complement cascade resulting in membrane lesioning, migration of leukocytes, natural killer cell migration, increased cellular permeability and subsequent tissue death. 5
Clinically the high specificity of NMO-IgG for the relapsing form of NMOSD optic neuritis, recurrent myelitis and a variety of encephalopathic presentations has been confirmed worldwide.
AQP4 antibody positivity is established as a diagnostic marker but it is significant in prognosticating the disease and for monitoring therapy. 8, 9 Our study included NMO IgG positive patients who were diagnosed using IF assay.
Blood/CSF analysis of AQP4 antibody is done using various biochemical methods. Its highly specific (>99%) and its sensitivity ranges from 48-72%. 6
MRI plays a pivotal role in diagnosis of NMOSD. In optic neuritis increased signal within the optic nerve may be seen with fat suppressed T2-weighted orbital MRI sequences and with gadolinium enhancement seen on T1-weighted sequences. Bilateral optic nerve involvement, posterior nerve predominance (especially with extension into the optic chiasm), or extensive lesions of the optic nerve more than half of its length are all suggestive of NMOSD.10 64% patients showed MRI evidence of NMO in the study population.
Treatment of NMOSD has two goals: suppression of acute inflammatory relapse and prevention of future relapses. High-dose intravenous methylprednisolone is adopted as the first-line agent to suppress inflammation in acute events followed by oral steroids tapered over 2–8 weeks on individual case severity. Other treatment modalities include plasmapharesis, immunosuppressants such as azathioprine, mycophenolate moefetil, mitoxantrone, etc. Although they have dose dependent side effects the risk to benefit ratio may warrant the use of suitable immunosuppressant for long term therapy in order to prevent relapse. 11,
Rituximab (RTX) is an anti-CD20 monoclonal antibody. 13 It acts by completely destroying a type of B-cell which has a downstream effect on rest of the immune system. 6
In 2005, Cree et al reported the first open-label study evaluating rituximab therapy in patients with NMO. In 2008, the beneficial effects of rituximab were reported in a retrospective multicenter study. 12 . It provides the best remission rates up of to 83 %. Additional doses of rituximab (375 mg/m2 × 4 total weekly doses or another 1000 mg, 2 weeks after the initial 1000 mg dose) provide long-lasting B cell depletion for 6–8 months. 11
Also CD27 (+) memory B cells assay are useful in determining the requirement for re-treatment and rituximab can be given if the count is more than 0.05% of peripheral blood mononuclear cells with 375/m2 of RTX-maintained remission.14
Multiple Sclerosis needs to be differentiated from NMO at the time of diagnosis in order to initiate effective specific NMO therapy and to prevent advocation of MS treatment for a case of NMO that may prove detrimental in many ways. In particular, interferon, fingolimod and natalizumab should be avoided as there is some evidence to suggest that these therapies may have a negative outcome or may even exacerbate NMOSD.1,15 In this study multiple sclerosis was ruled out clinically and by immunohistochemistry.
Financial constraints are are a major hurdle for diagnosis and treatment of this disabling condition.
CONCLUSION
Patients with NMO require a thorough assessment and correct diagnosis. NMO IgG antibody assay is a must for all patients presenting with optic neuritis with or without myelitis. Timely advocation of correct treatment and ensuring good rehabilitation may go a long way in preventing morbidity and supporting a smooth recovery. Immunosuppressants / long term steroids may play a role in prophylaxis. Rituximab therapy has shown promising results with wide safety range and should be considered in suitable patients.
REFERENCES
1.Broadley S, Khalili E, Heshmat S,Clarke L. Neuromyelitis Optica Spectrum Disorder. ACNR AUGUST-SEPTEMBER 2017; 17(1): 11-14.
2. Kim H J, Paul F,Tenembaum S, Asgari N, Palace J. MRI characteristics of neuromyelitis optica spectrum disorder. Neurology 2015; 84: 1165–1173.
3. Nandhagopal R, Abdullah Al-Asmi, Arunodaya R G. Neuromyelitis optica: an overview. Postgrad Med J 2010; 86: 153-159.
4. Wingerchuk DM, Lennon VA, Lucchinetti CF . The spectrum of neuromyelitis optica. Lancet Neurol 2007; 6: 805–15.
5. Jacob A, McKeon A, Nakashima I, Sato D K. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. Neurol Neurosurg Psychiatry 2013; 84: 922–930.
6. Mealy M A. Neuromyelitis Optica (NMO) and NMO spectrum disorder. The Transverse Myelitis Association Journal 2012; (1): 1-9.
7. Lennon V A, Kryzer T J, Pittock S J. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005; 202: 473–477.
8. Matiello M, Lennon VA, Jacob A. NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology 2008; 70: 2197–2200.
9. Weinshenker BG, Wingerchuk DM, Vukusic S. Neuromyelitis optica IgG predicts relapse after longitudinally extensive transverse myelitis. Ann Neurol 2006; 59: 566–569.
10. Wingerchuk DM, Banwell B, Bennett J L . International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85(2): 177-89.
11. Kessler R A, Mealy M A, Levy M. Treatment of Neuromyelitis Optica Spectrum Disorder: Acute, Preventive, and Symptomatic. Neurol 2016 January ; 18(1): 1-15.
12. Kim S H, Huh S-Y, Lee S J, Joung A, Kim H J. A 5-Year Follow-up of Rituximab Treatment in Patients With Neuromyelitis Optica Spectrum Disorder. JAMA Neurol 2013; 70(9): 1110-1117.
13. Joanna Kitley, BMBS; PatrickWaters, PhD; MarkWoodhall, PhD; M. Isabel Leite, DPhil; Andrew Murchison, BM BCh; . Neuromyelitis Optica Spectrum Disorders With Aquaporin-4 and Myelin-Oligodendrocyte Glycoprotein Antibodies A Comparative Study. JAMA Neurol 2014; 71(3): 276-283.
14. Kim SH, Kim W, Li XF. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol 2011; 68: 1412–20.
15. Bergamaschi R, Ghezzi A . Devic’s neuromyelitis optica: clinical features and prognostic factors. Neurol Sci 2004; 25: S364–S367.
APPENDIX
TABLE-1 BASELINE DEMOGRAPHIC CHARACTERISTICS
| S. No. | VARIABLE | PERCENTAGE (%) |
| 1. | Mean age +/- SD | 39.1 +/- 9.4 |
| 2. | Gender:
Male |
04 (25%) |
| Female | 12 (75%) |
TABLE-2 DESCRIPTIVE STATISTICS OF BCVA AT TIME OF PRESENTATION
| S. No. | BCVA ( LogMAR) | PERCENTAGE (%) |
| 1. | PL(Perception of Light) to 2.0 | 32% |
| 2. | 1.3-1.77 | 40% |
| 3. | 0.78-1.29 | 04% |
| 4. | 0.77-0.0 | 24%. |
TABLE-3 DESCRIPTIVE STATISTICS OF VISUAL OUTCOME FOLLOWING TREATMENT
| S. No. | BCVA ( LogMAR) | PERCENTAGE (%) |
| 1. | 2.0- PL | 32% |
| 2. | 1.30-1.77 | 40% |
| 3. | 0.78-1.17 | 4% |
| 4. | 0.47-0.77 | 16% |
| 5. | 0-0.30 | 8% |
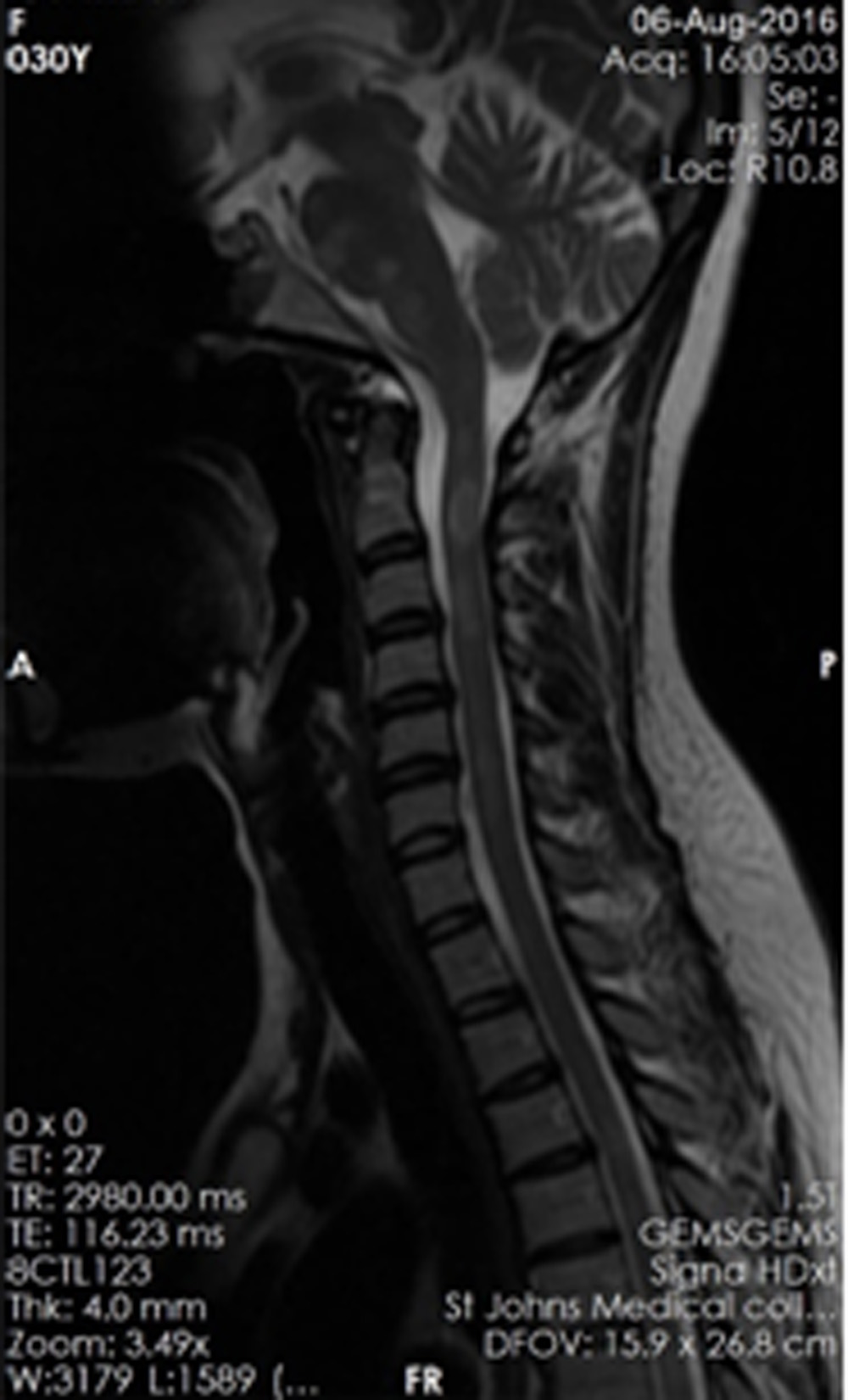
Multiple T2 hyperintense, intramedullary lesions extending over a long segment, predominantly involving the lateral and posterior cord are noted in the cervical cord from C1- C6 vertebral level. Some of these lesions were noted to show post contrast enhancement. The patient presented with mild weakness and paresthesia of the right arm.
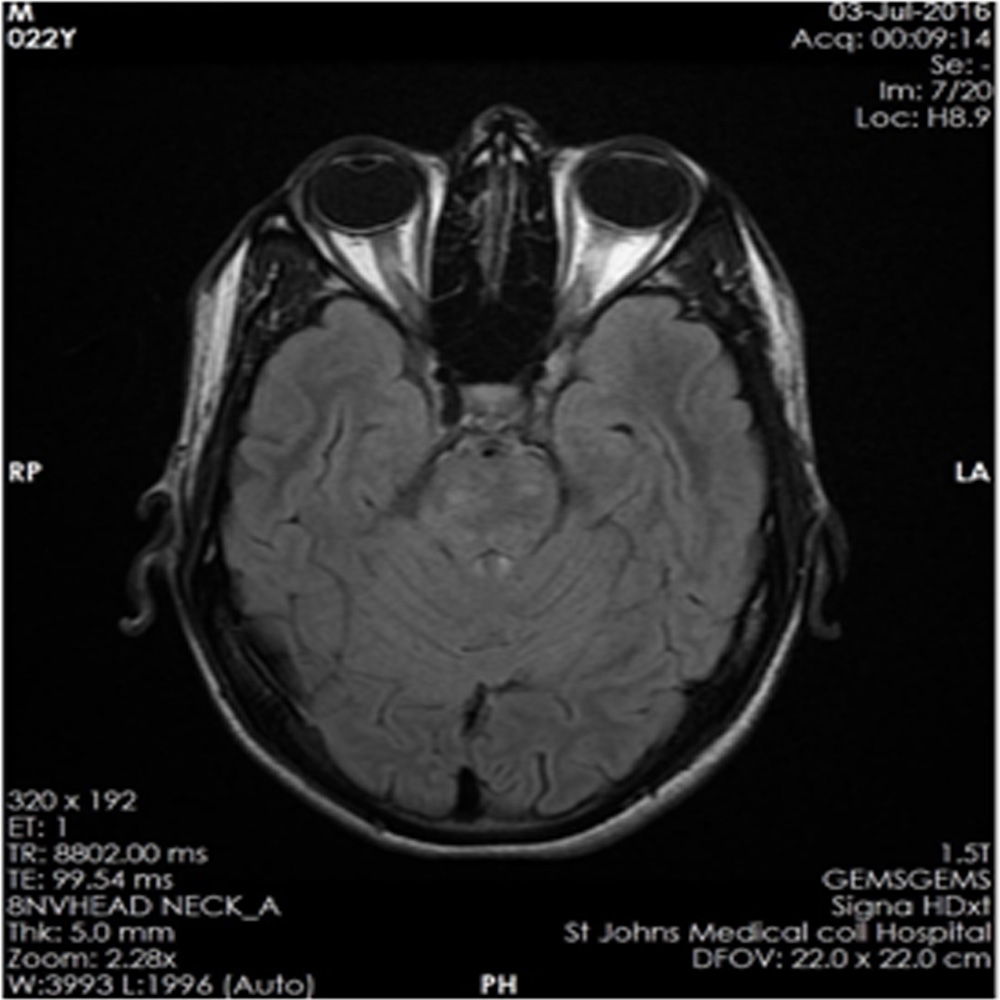
Nonspecific optic nerve sheath thickening noted on left side, optic nerve hyperintensities also noted along the intra-orbital course of optic nerve suggestive of optic neuritis. Patient also showed multiple thalamic lesions and cerebellar hyperintensities at various cut sections of MRI brain. The patient had presented with acute visual loss in left eye and vertigo
Leave a Comment